
|
EN NY INTELLIGENS – AI I DYRLÆGENS VERDENAI rykker ind. I en ny artikelserie stiller vi skarpt på, hvordan kunstig intelligens vinder indpas som redskab i det veterinære arbejdsfelt. Læs med og bliv klogere på, hvilke muligheder og udfordringer AI giver dig som dyrlæge. |


Campylobacteriose er den hyppigst rapporterede årsag til mave-tarminfektion i både Danmark og EU. Symptomerne omfatter typisk diarré, kvalme og feber, som er klassiske tegn på fødevare- eller vandbårne infektioner.
I Danmark toppede antallet af registrerede tilfælde i 2024 med over 5.500 rapporterede infektioner. Det faktiske antal anslås dog til at være op mod ti gange højere, da mange smittede ikke søger lægehjælp eller får stillet en diagnose (1). På EU-niveau blev der i 2023 rapporteret 148.181 tilfælde, svarende til en incidens på 45,7 tilfælde pr. 100.000 indbyggere. Til sammenligning var incidensen i Danmark samme år 87,6 pr. 100.000 (2).
Kyllingekød vurderes som den primære smittekilde, men også andre fødevarer som oksekød, drikkevand og kontakt med smittede dyr kan bidrage til smittespredningen.
Smittekilderegnskaber
For at opnå en dybere indsigt i smittevejene anvendes såkaldte smittekilderegnskaber, som estimerer, hvor mange humane sygdomstilfælde der kan tilskrives forskellige fødevare-, dyre- og miljøkilder. Disse estimater udgør et vigtigt beslutningsgrundlag for myndighederne i forhold til at målrette forebyggelses- og kontrolindsatser. Derudover kan metoden anvendes til at følge op på og evaluere effekten af implementerede tiltag over tid.
Grundprincippet bag smittekilderegnskaber er at opdele patogenet i undertyper og sammenligne fordelingen af disse mellem humane tilfælde og potentielle kilder ved hjælp af matematiske modeller. Denne tilgang har i mange år været anvendt af DTU Fødevareinstituttet – især i forbindelse med kontrol af Salmonella – og bygger på data fra fødevaremyndighedernes overvågning af dyr og fødevarer samt Statens Serum Instituts registreringer af humane tilfælde.
Metoden er løbende blevet videreudviklet i takt med fremkomsten af mere diskriminatoriske metoder til at inddele bakterier i undertyper, herunder fuld-genom sekventering (WGS), som har forbedret muligheden for at identificere smittekilder med større sikkerhed.
I modsætning til Salmonella er Campylobacter en mere kompleks og genetisk varieret mikroorganisme, hvilket har gjort det udfordrende at opdele bakterien i meningsfulde undertyper ved hjælp af traditionelle fænotypiske metoder. Tidligere er der anvendt multi-locus sequence typing (MLST), som klassificerer Campylobacter baseret på variation i syv såkaldte »husholdningsgener «. Erfaringen har dog vist, at denne metode ikke har tilstrækkelig opløsning til pålideligt at skelne mellem forskellige smittekilder.
I de senere år er WGS blevet en integreret del af den danske overvågning. Et Campylobacter-genom består af over 1,7 millioner basepar, og kernegenomet alene omfatter 1.343 loci. Denne mængde data giver en langt højere diskriminationsevne, men gør det samtidig praktisk umuligt at identificere undertyper manuelt uden bioinformatisk input og machine learning.
Siden 2019 er smittekilderegnskabet for Campylobacter blevet udarbejdet ved hjælp af machine learning (ML) modeller. ML er en underdisciplin inden for kunstig intelligens (AI), hvor algoritmer trænes til at identificere mønstre i data frem for at følge faste, foruddefinerede regler.
Metoden indebærer, at man typisk anvender 70 % af datasættet - bestående af Campylobacter-isolater fra fødevarer og dyr – til at træne algoritmen i at genkende genetiske signaturer, der er karakteristiske for forskellige smittekilder. Algoritmen lærer dermed at associere specifikke mønstre i genomerne med deres oprindelsesmiljø. Herefter valideres modellens præcision ved at teste den på de resterende 30 % af datasættet, hvor oprindelsen af isolaterne er kendt. Dette gøres for at vurdere, hvor præcist algoritmen kan forudsige smittekilden (Figur 1). Når modellen viser tilfredsstillende resultater, anvendes den på Campylobacter-isolater fra humane tilfælde, hvor den faktiske smittekilde er ukendt. Outputtet er sandsynligheden for, at et givent humant Campylobacter-tilfælde stammer fra en specifik smittekilde (3).
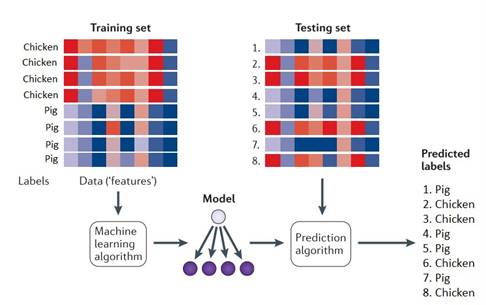
Figur 1 – Princippet for smittekilderegnskabsmodellen baseret på Machine Learning (ML): ML-modellen er baseret på et trænings- og testsæt, hvor fødevarekilden er kendt. På figuren ser man et eksempel med overvejende røde labels for kylling og blå for svin. Det er disse mønstre, som modellen finder i datasættet, og som bruges til at forudsige smittekilden for humane Campylobacter-infektioner.
Campylobacter på tværs af grænser
Tidligere i år publicerede vi en videnskabelig artikel, der netop benytter machine learning til at udarbejde et smittekilderegnskab for humane Campylobacter-infektioner på tværs af forskellige EU-lande (4). Igennem et EU-forskningsprojekt »Discovering the sources of Salmonella, Campylobacter, VTEC and antimicrobial resistance (DiSCoVer)« indenfor One Health European Joint Program (OHEJP), blev der indsamlet WGS-data fra Campylobacter-isolater fra otte lande (Danmark, Frankrig, Irland, Holland, Polen, Portugal, Spanien og Sverige).
Antallet af prøver taget og isolater i alt og pr. smittekilde varierede fra land til land. Et af formålene med studiet var derfor også at undersøge, hvordan prøvetagningsstrategier og stikprøvestørrelser kan påvirke nøjagtigheden af smittekildemodellernes forudsigelser.
Materialer og metoder
I alt havde vi WGS-data fra 6.313 Campylobacter-isolater, heraf 2.627 fra mennesker med Campylobacter-infektion og 3.686 prøver fra diverse smittekilder (fx ferskvand, produktionsdyr, kæledyr, vilde dyr og fødevarer). Antallet af prøver varierede fra 90 til 2.846 pr. land, hvor Danmark bidrog med det største antal prøver og Sverige med det mindste.
Datasættet var skævt fordelt med en overvægt af prøver fra slagtekyllinger, især fra Danmark (829 prøver), Irland (272 prøver) og Holland (254 prøver), mens andre kilder var underrepræsenterede, som fx prøver fra kat og vilde dyr. Frankrig og Portugal havde indsamlet prøver fra en forholdsvis bred vifte af kilder, men med relativt få prøver fra hver kilde. Omvendt bidrog lande som Irland og Polen med et større antal prøver, men fra et mere begrænset antal kilder. Danmarks datasæt omfattede også et
betydeligt antal prøver fra importeret fjerkræ. Frankrig og Sverige havde ingen prøver fra mennesker tilgængeligt.
For at definere smittekildekategorierne blev prøverne grupperet efter land, oprindelse/import og dyreart/reservoir. Det vil sige, at prøver fra henholdsvis levende slagtekyllinger og kyllingekød blev grupperet i samme kategori: Slagtekyllinger. Det samme gjorde sig gældende for kvæg/oksekød, gris/svinekød osv. Vi valgte også at gruppere prøver fra får og geder på grund af de lave antal prøver fra disse dyrearter/fødevarer.
For at estimere smittekilderne til de humane tilfælde benyttede machine learning-modellen frekvensen af såkaldte k-mers. En k-mer er en kort DNA-sekvens af en bestemt længde. »k« angiver antallet af baser, man har i DNA-sekvensen. Modellen beregner, hvor mange der findes af hver k-mer sekvens i genomet og bruger denne frekvensfordeling til at forudsige smittekilden under antagelse af, at jo større lighed, der er i frekvensfordelingen, jo større er sandsynligheden for, at isolaterne kommer fra samme kilde.
Nærværende model var baseret på k = 9, dvs. optælling af sekvenser bestående af ni baser, som fx »ATGCGATTC«. Den endelige model anvendte i alt 277 forskellige 9-mer sekvenser til at finde genetiske mønstre, der kunne bruges til at skelne mellem de forskellige smittekilder. Resultaterne angiver sandsynligheden for, at et givent humant Campylobacter-tilfælde stammer fra en specifik smittekilde. Kun tilfælde med en sandsynlighed på over 70 % for at komme fra en specifik kilde medtages. Smittekilden til tilfælde med sandsynligheder under 70 % betegnes som »ukendt kilde«.
For at undersøge, hvordan stikprøvestørrelser påvirker modellens præstation, blev der udført både up- og down-sampling af datasættet. Up-sampling blev anvendt for at balancere datasættet ved at kopiere antallet af prøver i underrepræsenterede kilder, så prøveantallet matchede dem i den største kategori. Smittekilder med mindre end 50 prøver blev således up-samplet til det største antal prøver. Fx blev »and/andekød« up-samplet fra 4 til 829 i det danske datasæt.
I en anden model blev down-sampling udført for at reducere antallet af prøver i overrepræsenterede kategorier, så de matchede størrelsen af en mindre kategori. Kilder med over 300 prøver blev down-samplet for at opnå en bedre balance i datasættet. Som et eksempel herpå blev »slagtekyllinger« reduceret fra 829 til 300 prøver i det danske datasæt. Ved down-sampling undgår man kunstigt at øge antallet af prøver, som det gøres med up-sampling. Resultaterne af de to modeller blev derefter sammenlignet.
Resultater
Ved hjælp af up-sampling-modellen blev slagtekyllinger fra Danmark udpeget til at være den vigtigste smittekilde for danske tilfælde (199/232 tilfælde, 86 %), efterfulgt af kvæg/oksekød fra Danmark (19/232 tilfælde, 8 %). Kun få danske humane tilfælde kunne tilskrives kilder uden for Danmark, såsom importeret slagtekyllinger (6/232 tilfælde, 3 %), kød fra slagtekyllinger i Polen (2/232 tilfælde, 1 %) eller Holland (2/232 tilfælde, 1 %). Modellen estimerede, at ni humane tilfælde fra andre lande (Holland, Portugal, Irland) var blevet smittet af danske slagtekyllinger (9/41, 22 %) (Figur 2).

Figur 2 - Resultaterne af den up-samplede smittekilderegnskabsmodel: Kun humane Campylobactertilfælde (N=273) med over 70 % sandsynlighed for at stamme fra en specifik smittekilde er vist i diagrammet. Labelforklaring: DK=Danmark, NL=Holland, ES= Spanien, PL=Polen, PT=Portugal, IE=Irland, FR=Frankrig, IMP=Importeret, Broiler=Slagtekylling, Cattle=Kvæg, Pig=Gris, Dog=Hund.
Ved at bruge det down-samplede datasæt blev humane tilfælde fra Danmark overvejende tilskrevet danske slagtekyllinger (226/365 tilfælde, 62 %), mens nogle også blev tilskrevet dansk kvæg/oksekød (54/365 tilfælde, 15 %) samt slagtekyllinger fra Holland (22/365 tilfælde, 6 %). Omkring en femtedel af danske tilfælde blev tilskrevet udenlandske kilder (75/365 tilfælde, 21 %). Den down-samplede model estimerede, at 50 humane tilfælde fra andre lande (Holland, Portugal, Irland, Spanien) stammede fra danske slagtekyllinger (53/170, 31 %) (Figur 3).

Figur 3 - Resultaterne af den down-samplede smittekilderegnskabsmodel: Kun humane Campylobactertilfælde (N=535) med over 70 % sandsynlighed for at stamme fra en specifik smittekilde er vist i diagrammet. Labelforklaring: DK=Danmark, NL=Holland, ES= Spanien, PL=Polen, PT=Portugal, IE=Irland, FR=Frankrig, SE=Sverige, IMP=Importeret, Broiler=Slagtekylling, Cattle=Kvæg, Sheep_goat= Får_ged, Pig=Gris, Turkey=Kalkun, Duck=And, Dog=Hund, Wild_bird=Vilde fugle, Freshwater=Fersk vand.
Overordnet viser resultaterne, at ved brug af den up-samplede model, blev der generelt tilskrevet flere tilfælde til smittekilder med mange prøver sammenlignet med smittekilder med færre prøver. Fx blev 86 % af danske humane tilfælde tilskrevet kylling i den up-samplede model sammenlignet med 62 % i den down-samplede model.
For kvæg/oksekød forholdt det sig omvendt med 8 % i den up-samplede model mod 15 % i den down-samplede model. I den down-samplede model blev flere kildekategorier desuden udpeget som smittekilde til humane infektioner, ligesom modellen kunne angive en kilde for flere af de humane infektioner (535 vs. 273).
Udover disse to modeller, som inkluderer data fra alle de otte lande, udarbejdede vi også individuelle landemodeller for henholdsvis Danmark, Irland, Holland, Polen, Portugal og Spanien. Resultaterne er ikke medtaget i denne artikel, men de bekræfter, at kylling i alle lande fremstår som den vigtigste smittekilde.
Diskussion
Resultaterne fra ML-modellen indikerer, at langt størstedelen af humane Campylobacter-infektioner kan relateres til fødevareproducerende husdyr. Dette stemmer overens med tidligere studier, der fremhæver husdyr som et væsentligt smittereservoir. Selvom slagtekyllinger fortsat fremstår som den primære kilde til humane infektioner, viser vores resultater, at modellerne potentielt kan overvurdere betydningen af kilder, der er overrepræsenterede i datasættet. Denne skævhed kan føre til en form for selvopfyldende profeti. Når en bestemt kildekategori som fx slagtekyllinger er omfattende prøveudtaget, introduceres der mere genetisk variation i datagrundlaget for netop denne kilde. Det øger sandsynligheden for, at humane isolater matches til den. Omvendt kan kilder med færre prøver fremstå som mindre betydningsfulde, da den genetiske variation i disse grupper er mere begrænset, hvilket reducerer chancen for korrekt allokering af humane tilfælde.
I dette studie anvendte vi k-mers med længde 9 (k=9) til at identificere mønstre i Campylobacter-genomer fra forskellige smittekilder. På trods af modellens relativt høje nøjagtighed blev en betydelig andel af de humane tilfælde allokeret til »ukendt kilde«, hvilket sker, når sandsynligheden for en tilknytning til en specifik kilde er under 70 %. Dette kan indikere, at Campylobacter-stammer er udbredt på tværs af flere kilder, hvilket udfordrer modellens evne til at skelne mellem dem.
En højere k-værdi kan potentielt forbedre modellens diskriminatoriske evne, men dette giver nogle udfordringer. For med en øget k-mer længde, vokser antallet af mulige kombinationer for hver af de fire nukleotider (A, C, G, T) eksponentielt, hvilket fører til beregningsmæssige begrænsninger. Samtidig bliver længere k-mers mere specifikke og dermed sjældnere, hvilket begrænser data om k-mer-frekvenser på tværs af prøver. Omvendt er kortere k-mers mere modtagelige for sekventeringsfejl, hvilket kan forstyrre analysens nøjagtighed. Det er derfor afgørende at anvende genomer af høj kvalitet for at sikre en pålidelig identifikation af genetiske mønstre på tværs af kilder.
Den optimale brug af k-mer kræver en balance mellem sensitivitet og robusthed, og på baggrund af disse hensyn blev 9-mers vurderet som den mest hensigtsmæssige længde i dette studie.
I tidligere studier har vi også anvendt og sammenlignet alternative bioinformatiske inputmetoder, herunder coregenome MLST (cgMLST), hvor 1.343 loci benyttes til at klassificere Campylobacter-undertyper (3). En væsentlig udfordring ved cgMLST er dog, at der ofte forekommer manglende alleldata, som efterfølgende estimeres baseret på de øvrige prøver i datasættet. Denne imputering introducerer yderligere usikkerhed i de endelige resultater. På baggrund af vores erfaring vurderer vi derfor, at k-mer-baserede analyser til trods for deres beregningsmæssige kompleksitet tilbyder en mere robust og pålidelig tilgang til kildesporing.
Dette studie peger på en central udfordring ved brug af WGS-data i smittekilderegnskaber for fødevarebårne sygdomme: Skæve og ufuldstændige datasæt kan føre til misvisende vurderinger af de enkelte smittekilders betydning. For at opnå mere pålidelige smittekildeestimater bør overvågningsprogrammer stræbe efter en mere balanceret og repræsentativ prøveudtagning på tværs af relevante kilder. Dette kræver en styrket One Health-tilgang, hvor prøver fra mennesker, fødevarer, dyr og miljø analyseres samlet og standardiseret. En sådan strategi, tilpasset nationale prioriteter og ressourcer, vil styrke grundlaget for effektive kontrol- og forebyggelsestiltag.


