
Patienter i status epilepticus er en akutmedicinsk patientgruppe, som alle klinikkere bør være forberedt på, da netop denne situation sætter store krav til klinikerens akutberedskab og paratviden. Klinikeren står med mange bolde i luften, og tidspresset er stort. I sådanne situationer er det hjælpsomt at have en protokol ved hånden, som den der beskrives i denne artikel.
Status epilepticus (SE) defineres som gentagne anfald imellem hvilke, patienten ikke genvinder fuld bevidsthed, eller som varer mere end 5 minutter (1-6). I tabel 1 ses en liste over de kliniske definitioner relateret til anfaldspatienter.
Årsagssammenhæng
SE kan enten have epileptisk oprindelse (idiopatisk eller strukturel epilepsi) eller opstå som følge af non-epileptiske, reaktive anfald med ekstrakraniel ætiologi (såsom intoksikation) som vist i figur 1. Med andre ord; betegnelsen SE dækker over ethvert ikke-selvbegrænsende anfald af over 5 minutters varighed, uanset ætiologi.
Definitioner vedrørende anfald hos hund |
|
| Anfald (eng. Seizure) | Enhver pludselig, selvbegrænsende og abnorm episode. Omfatter ikke kun epileptiske anfald. |
| Epileptisk anfald | Overdreven, synkron og oftest selvbegrænsende elektrisk aktivitet (epileptisk aktivitet) i hjernens neuroner. Udtrykkes klinisk som anfald med motoriske (konvulsive), autonome og/eller adfærdsrelaterede tegn. Kan være fokale eller generaliserede. |
| Krampeanfald | Tonisk/kloniske anfald. |
| Fokale anfald | Anfald, som udspringer af fokal/regional abnorm elektrisk aktivitet i hjernen. Udtrykkes klinisk som anfald med motoriske, autonome og/eller adfærdsrelaterede tegn. Ved motoriske anfald ses lateralisering. |
| Generaliserede anfald |
Anfald som udspringer fra samtidig bilateral abnorm elektrisk aktivitet i hjernen. Udtrykkes klinisk overvejende som krampeanfald med bevidsthedstab, men kan i meget sjældne tilfælde forekomme som anfald med serier af ultrakort bevidsthedssvigt, der viser sig som fjernhed (absencer). |
| Epilepsi | En diagnose karakteriseret ved kortvarige, gentagne, stereotypt optrædende anfald forårsaget af forstyrret elektrisk aktivitet i hjernen. Der skal have været minimum 2 anfald med mere end 24 timers mellemrum for at opfylde det kliniske kriterie for epilepsi. |
| Nonepileptiske reaktive anfald |
Anfald, der er fremprovokeret af en ekstrakraniel årsag. Der er her tale om hjernens naturlige respons på en udefra kommende systemisk påvirkning, f.eks. en metabolisk forstyrrelse eller forgiftning. Denne situation er reversibel, hvis underliggende årsag korrigeres. |
| Strukturel epilepsi | Epilepsi, hvor anfald opstår som følge af påviselig intrakraniel patologi (fx kongenital abnormalitet, neoplasi, inflammatorisk CNS sygdom eller stroke) |
| Idiopatisk epilepsi |
Epilepsi, hvor anfald opstår som følge af en genetisk eller mistænkt genetisk ætiologi, samt anfald hvor de underliggende mekanismer for anfaldets opståen ikke er erkendt, men hvor der formentlig ligesom for de genetiske epilepsier er tale om ubalancer af neurotransmittersystemer (typisk GABA/glutamat) eller dysfunktion af ionkanaler. Ved idiopatisk epilepsi er de epileptiske anfald det eneste tegn på sygdom og patienten er klinisk og neurologisk normal interiktalt. En strukturel årsag til anfald (strukturel epilepsi) er afvist. |
| Status epilepticus | Gentagne anfald imellem hvilke patienten ikke genvinder fuld bevidsthed ELLER anfald som varer mere en 5 minutter. Uagtet ætiologi, dvs. både intra- og ekstrakraniel oprindelse. |
| Refraktær status epilepticus |
Pågående status epilepticus som ikke responderer på adækvat behandling med benzodiazepin, phenobarbital, og anden passende akutmedicinsk antiepileptisk behandling. |
| Klyngeanfald (eng. Cluster seizures) |
Ophobede anfald, 2 eller flere indenfor 24 timer, hvorimellem patienten genvinder normal bevidsthed. |
Tabel 1. Definitioner vedrørende anfald hos hund (6).
SE opstår, når de mekanismer, der normalt sørger for, at anfald er selvbegrænsende og kortvarige, svigter. Den voldsomme anfaldsaktivitet ved SE vedligeholdes af overdreven og vedvarende elektrisk fyring fra større eller mindre grupper af neuroner i hjernen (1,4-7).
SE klassificeres i to undertyper: Generaliseret konvulsivt SE og fokalt SE. Generaliseret SE kendes som de typiske toniske, kloniske eller tonisk-kloniske generaliserede motoranfald (krampeanfald), som involverer neuroner udbredt i hele hjernen, og hvor der sædvanligvis er tab af bevidsthed.
Den kliniske manifestation af fokalt SE afspejler derimod den anatomiske lokalisation i hjernen, hvorfra der er lokaliseret anfaldsaktivitet. Det kan være som fokale motoriske anfald med muskeltrækninger eller ryk i fx ansigtet eller en enkelt ekstremitet, autonome fænomener som hypersalivation og pulpildilatation eller adfærdsrelaterede kliniske tegn. Disse kan forekomme alene eller i kombination.
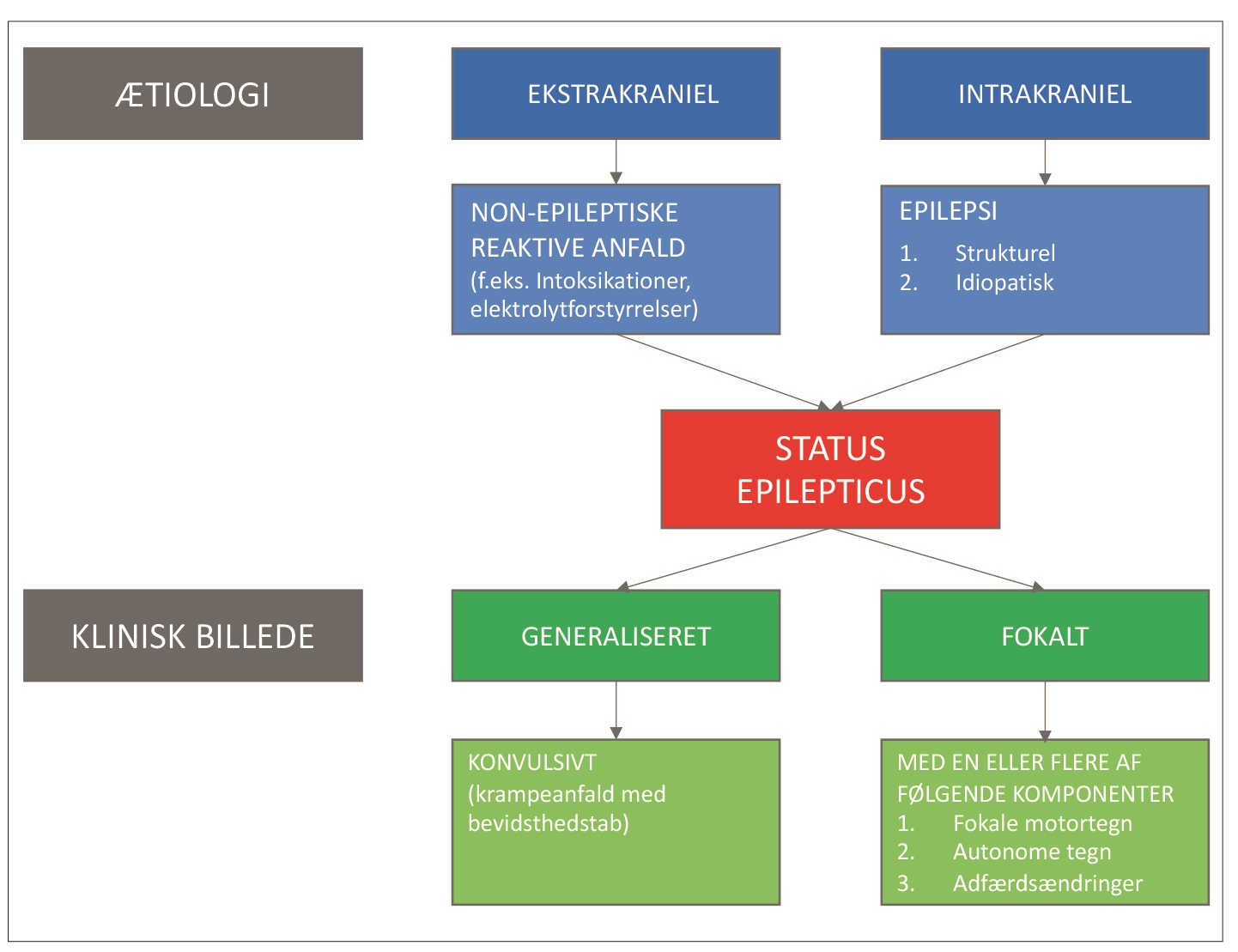
Figur 1. Klassificering af status epilepticus (SE) på baggrund af henholdsvis ætiologi og klinisk billede. Bemærk, at både ekstra- og intrakranielle sygdomstilstande kan give anledning til SE, og at SE kan komme til udtryk som både generaliserede og fokale anfald (6).
Selvom fokalt SE kan se mere fredeligt ud end generaliseret konvulsivt SE, er begge farlige for hjernen og kræver derfor også begge stor bevågenhed og hurtig indgriben. I øvrigt kan fokalt SE udvikle sig til generaliseret konvulsivt SE, hvilket man skal undgå. Endelig kan der ved behandling af det generaliserede konvulsive SE opstå den situation, at man har stoppet de generaliserede kramper, men at der stadig forekommer fokal anfaldsaktivitet, fx i form af tics i ansigtet. I så fald er nogle af hjernens neuroner stadig hyperaktive og status dermed ikke under kontrol (1, 5, 8).
Såkaldte klyngeanfald, karakteriseret ved, at patienten mellem anfald genvinder normal bevidsthed, skal ikke forveksles med SE. Patienter med klyngeanfald er dog også en kritisk patientgruppe, som kræver akut behandling og ofte også indlæggelse (1, 5-11).
I tabel 2 ses en sammenligning af tre studier (alle retrospektive), som har undersøgt årsager til SE (epileptiske og non-epileptiske). Omkring 27-38 % af patienterne har idiopatisk epilepsi som underliggende årsag, mens stort set det samme antal patienter (32-40 %) har en underliggende strukturel årsag, såsom hjernetumor eller inflammatorisk hjernesygdom (3, 7, 12).
Klassificering af status epilepticus på baggrund af ætiologi og prævalens |
|||
| Bateman & Parent, 1999 | Platt & Haag, 2002 | Zimmermann et al., 2009 | |
| Antal cases (n) | 194 | 50 | 88 |
| Ætiologi | |||
| Idiopatisk epilepsi, % | 27 | 28 | 38 |
| Strukturel epilepsi, % | 35 | 32 | 40 |
| Meningoencefalitis, % | 23 | 12 | 15 |
| Neoplasi, % | 4 | 12 | 24 |
| Andre (inkl. traume og stroke), % | 9 | 8 | 1 |
| Reaktive anfald, % | 7 | 12 | 23 |
| Metabolisk årsag, % | 5 | 4 | 8 |
| Intoksikation, % |
2 | 8 | 15 |
| Andre, % |
31 | 28 | |
| Lav serumkoncentration af antiepileptisk medicin, % |
6 | N/A | |
| Uden kendt ætiologia, % |
25 | 28 | N/A |
| aHvor hverken intoksikation eller idiopatisk epilepsi synes sandsynligt |
|||
Tabel 1. Definitioner vedrørende anfald hos hund (6).
Studiet af Bateman og Parent (1999) undersøgte 194 hunde med SE eller klyngeanfald, hvoraf 49 % og 25 % allerede var kendt med epilepsi og i peroral antiepileptisk behandling med henholdsvis phenobarbital eller kaliumbromid ved indlæggelsen. Samme studie fandt, at ca. 6 % af indlagte patienter med SE havde for lav serumkoncentration af epilepsimedicin (7), hvilket kunne være medvirkende årsag til udviklingen af SE. Dette understreger vigtigheden af kontinuerlig monitorering af medicinering hos epilepsipatienter.
Det er i de tre studier fundet, at der hos 2-15 % af patienterne i SE eller med klyngeanfald var tale om en intoksikation som underliggende årsag (3, 7, 12). Denne diagnose bygger traditionelt på de anamnestiske oplysninger eller på direkte beviser som eksempelvis en drugtest. Kategorien »ukendt ætiologi« i disse studier dækker over en patientgruppe, hvor hverken intrakraniel eller ekstrakraniel årsag kunne påvises, og hvor sygdomsforløbet og patientens signalement ej heller var foreneligt med idiopatisk epilepsi. Man kan mistænke, at denne gruppe måske omfatter flere uerkendte intoksikationer.
Forekomst
Den sande forekomst af SE kendes ikke, men der er estimeret en prævalens af SE på mellem 0,44 % og 0,77 % blandt patientpopulationer fra henvisningshospitaler (3,7). Man ved dog, at der hos epilepsipatienter er en øget risiko for at opleve status epilepticus, hvilket man derfor altid bør informere ejere til disse patienter om. Et retrospektivt studie omhandlende 78 hunde med idiopatisk epilepsi og 24 hunde med strukturel epilepsi fandt, at henholdsvis 18 % og 25 % af disse oplevede SE (13), hvilket ikke overraskende viser en større risiko for SE ved strukturel epilepsi.
Et andet retrospektivt studie inkluderende 407 hunde med idiopatisk epilepsi har dog fundet, at kun 2,5 % udviklede SE (14). Det er desuden dokumenteret, at race kan have indflydelse på risikoen for at udvikle SE. Dette skal ses i lyset af visse racers arvelige prædisponering for behandlingsrefraktær idiopatisk epilepsi, som det fx ses hos border collie og australian shepherd (15, 16), eller for udvikling af strukturel epilepsi som følge af primære hjernesygdomme som fx inflammatorisk hjernesygdom eller hjernetumor (2, 6, 7, 17, 18).
Anfaldsfysiologi, kompensatoriske mekanismer og faser af SE
Alle individer har en individuel anfaldstærskel, som kan være påvirket af forskellige faktorer, herunder genetiske, og som har betydning for bl.a. neuronernes miljø. Anfald opstår, når miljøet forstyrres, og anfaldstærsklen sænkes.
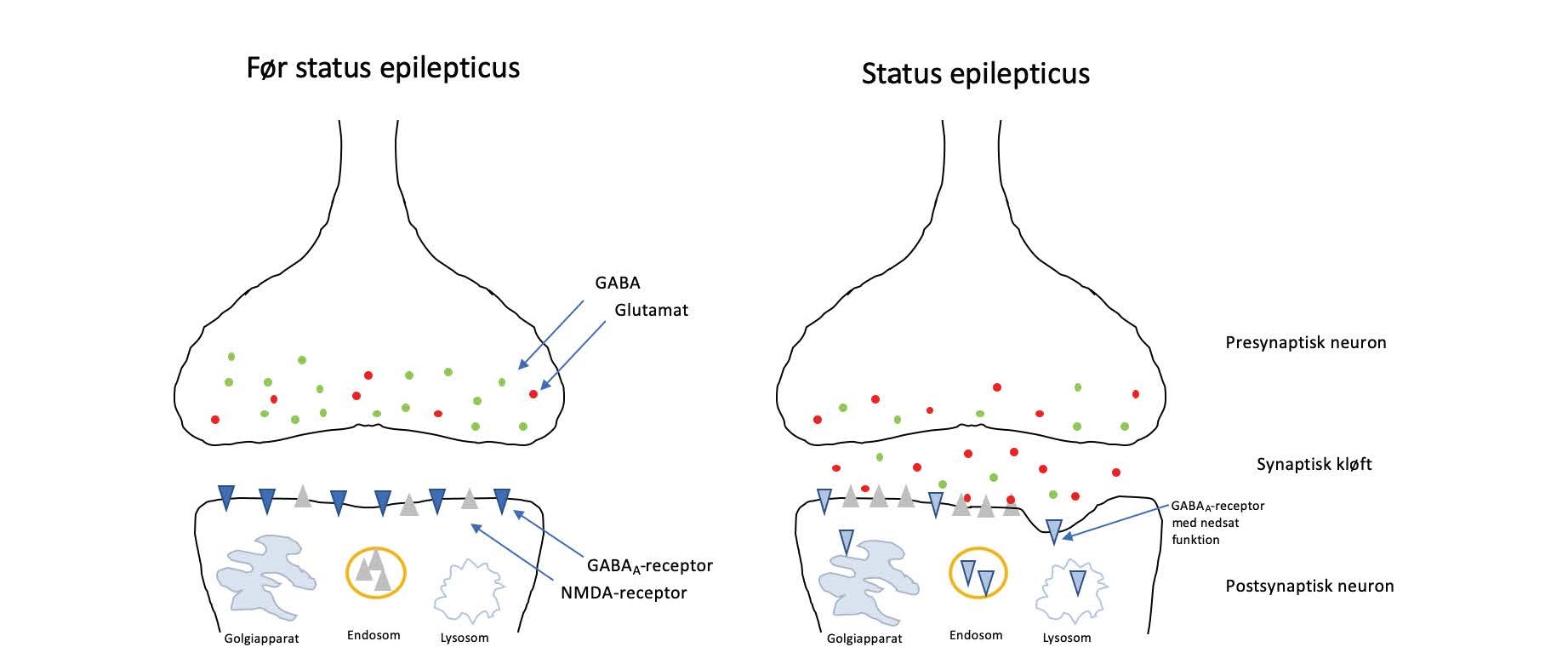
Figur 2. Skematisk illustration viser nedreguleringen af GABAA-receptorer, samt opregulering af NMDA-receptorer ved status epilepticus. Efter langvarig status epilepticus (>30min) kompromitteres de inhiberende GABAA-receptorer, i takt med at tilstedeværelsen af excitatorisk glutamat stiger, og antallet af aktive NMDA-receptorer på postsynaptiske membran opreguleres (modificeret efter Nair et al., 2011 (22)). GABA = gamma-aminobutyric acid; NMDA = N-methyl-D-aspatate.
To neurotransmittere spiller en særlig stor rolle i udvikling og afslutning af anfald; nemlig inhibitorisk GABA (gamma-aminobutyric acid) og excitatorisk glutamat samt deres receptorer, som primært er hhv. GA-BAA- og N-methyl-D-aspartate (NM-DA)-receptorer (1, 10, 15, 19-22).
Hjernens egen autoregulation kan ved SE indledningsvis opretholde en succesfuld regulering af cerebral ilt-tilførsel, glukoseniveau og tryk. Men allerede efter 5 minutter kommer hjernens normalfysiologiske mekanismer under pres, og efter ca. 30 minutters SE begynder denne regulering at svigte. Langt de fleste anfald er som tidligere nævnt selvbegrænsende, men de præcise homøostatiske mekanismer bag afslutning af anfald kendes ikke, og dermed er mekanismerne bag udviklingen af SE heller ikke fuldt afklarede (1, 20, 21). Man ved, at: 1) der fra starten af SE sker en løbende nedregulering og reduceret funktion af GABAA-receptorer og dermed en gradvis nedsat inhibitorisk effekt; 2) der sker en opregulering af NMDA-receptorer og øget frigivelse af glutamat efter 30 minutter og dermed en øget excitatorisk effekt; 3) øget glutamat- og NMDA-aktivitet er forbundet med neuronal celledød, og dermed opstår der risiko for irreversibel hjerneskade efter 30 minutters SE (1, 5, 10, 19, 23). Se i øvrigt figur 2 og tabel 3.
De fleste førstevalgs antiepileptika (benzodiazepiner og barbiturater), som bruges både humant og veterinært, virker ved at fremme GABA-receptorernes inhibitoriske effekt (se figur 3) (1, 3-5). Eftersom disse receptorer nedreguleres løbende, er det optimale behandlingsvindue derfor begrænset til de første 30 minutter af SE.
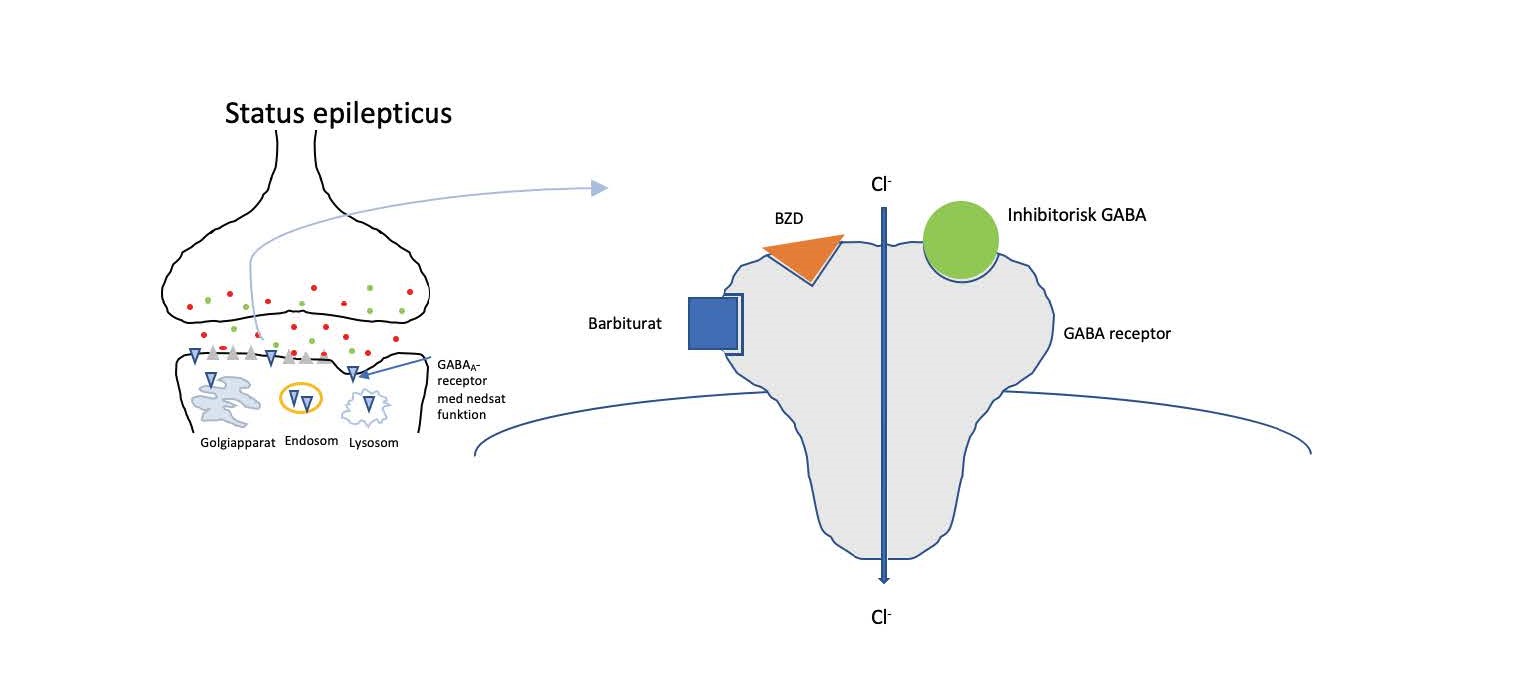
Figur 3. Den inhibitoriske GABAA-receptor stimuleres af GABA. Både benzodiazepiner og barbiturater er netop gabaerge og stimulerer denne receptor, dog på forskellig vis. Barbiturater og benzodiazepiner er førstevalg i behandling af status epilepticus. BZD = benzodiazepin; GABA = gamma-aminobutyric acid.
Udover ændringerne i hjernens lokale miljø, som sætter hjernen under stort pres ved vedvarende SE, kommer også kroppens vitale funktioner i fare. I takt med, at hjernens overordnede regulering svigter, udvikles en række systemiske, kompensatoriske mekanismer, som har betydning for klinikerens tilgang til patienten. Man taler om tre faser af SE: 1) en kompensatorisk fase, 2) en dekompensatorisk fase og 3) en refraktær fase, dvs. behandlingsrefraktær fase. Denne faseinddeling er tæt associeret med behandlingsvinduet og prognosen for at restituere patienten (se tabel 3).
Indledningsvist forekommer systemisk hypoksi, men hjernens autoregulation imødekommer dette i den kompensatoriske fase gennem systemisk vasodilatation. Også hyperglykæmi forekommer i den kompensatoriske fase, hvor der bliver rekrutteret ekstra glukose til hjernens øgede energiforbrug. Senere i forløbet bliver kroppens depoter udtømt, og der opstår systemisk hypoglykæmi. Herudover er der på grund af den vedvarende muskelaktivitet risiko for udvikling af hypertermi, hvorfor patientens temperatur skal følges nøje, og afkølende foranstaltninger iværksættes ved behov (3-5, 9, 10, 20, 21). Allerede fra starten af SE sker der en tiltagende øget frigivelse af catecholaminer, som systemisk og over tid medvirker til bl.a. takykardi og hypertension. Patienten bliver desuden respiratorisk kompromitteret som følge af autonom og central påvirkning. Dette sammenholdt med den høje neuronale og muskulære aktivitet fører til systemisk acidose, der forværres med anfaldets længde. Som følge af de ovenfor beskrevne mekanismer er den kompensatoriske fase også klinikerens behandlingsvindue, altså indenfor 30 min.
På grund af de tiltagende komplikationer i hjernen og systemisk er det en stor udfordring at stabilisere patienten i den dekompensatoriske fase. Risikoen for udvikling af hjerneødem og eventuelt herniation, som kræver akut handling, er overhængende i denne fase. Når patienten kommer i den refraktære fase er det fatalt, og patientens tilstand er ofte så dårlig, at der må vælges aflivning (se tabel 3).
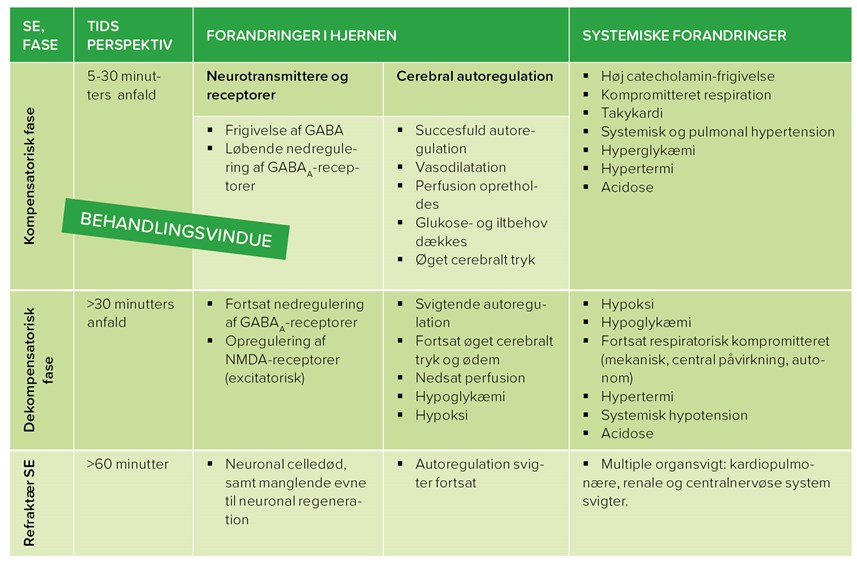
Tabel 3. De 3 faser af status epilepticus i forhold til tid, samt den konkrete udvikling i hjernen og systemisk. Det afgørende behandlingsvindue for status epilepticus strækker sig over de første 30 minutter af anfaldet. Refraktær status epilepticus ses ved manglende effekt af first-line behandling med benzodiazepin og phenobarbital. GABA = gamma-aminobutyric acid; NMDA = N-methyl-D-aspatate.
Håndtering af SE-patienter
De tre vigtigste komponenter i håndteringen af SE-patienter er: 1) opretholdelse af vitale funktioner, 2) opnåelse af anfaldskontrol ved at standse den øgede elektriske aktivitet i hjernen og 3) identificering og behandling af evt. underliggende årsag (3-5, 9, 10, 20-24).
Opretholdelse af vitale funktioner under SE
En patient i SE er først og fremmest en akutmedicinsk patient. Det, der foregår i hjernen, og som for klinikeren er synligt som vedvarende anfaldsaktivitet, er kun toppen af isbjerget. Derfor er risikoen for at miste patienten stor, hvis man undlader at undersøge og håndtere det, som foregår under overfladen. Håndteringen af SE-patienter kræver kapacitet til intensiv behandling og monitorering.
Første skridt i håndteringen af SE-patienter er at sikre vitale funktioner, herunder frie luftveje, tildeling af ilt, samt skaffe IV-adgang. Hypoksi, hypotension og hypertermi korrelerer i lige så høj grad med omfanget af neuronal skade som længden af anfaldet (24). Derfor anbefales løbende monitorering af blodtryk, temperatur og iltmætning, samt syre-base-status. Der bør anlægges intravenøst kateter til brug for væsketerapi, og dermed opretholdelse af blodtrykket, samt til behandling med intravenøs antiepileptika.
Alle SE-patienter bør tildeles ilt med iltmaske. Det er i øvrigt vigtigt at være klar til eventuel intubering i fald, der ses markant respirationsdepression eller mekanisk blokering af luftveje, hvilket kan være ekstra relevant for brachycephale hunde. Desuden virker de fleste antiepileptika dæmpende på respirationen, hvilket betyder, at der skal være ekstra opmærksomhed på behovet for intubering og ilt. En syre-base-måling kan give information om patientens pH, CO2-mætning, ilt-mætning (kræver ideelt set en arteriel blodprøve), samt bicarbonat-koncentration. Oftest ses en respiratorisk acidose, eventuelt med sekundær kompensatorisk metabolisk respons, som ses ved øget bicarbonat. Herudover bidrager muskelaktiviteten til den metaboliske acidose, idet der frigives laktat. To vigtige tommelfingerregler er, at en pH under 7,2 er livstruende, og tildeling af intravenøs bicarbonat ved respiratorisk acidose er kontraindikeret. Derfor er adækvat ventilation og ilttilførsel samt væsketerapi klinikerens vigtigste værktøjer i denne sammenhæng.
Blodglukose hos patienter i SE vil som tidligere beskrevet være høj i begyndelsen af forløbet pga. høj catecholaminfrigivelse med rekruttering af nødvendig glukose til hjernens øgede forbrug (med undtagelse af SE udløst af hypoglykæmi). Senere i forløbet, hvor depoterne er tømt, kan der opstå regulær behandlingskrævende hypoglykæmi. Opmærksomheden henledes dog på, at der fra eksperimentelle dyrestudier er tegn på, at hyperglykæmi øger risikoen for neuronal skade (1). Derfor bør måling af glukose også altid gå forud for intravenøs behandling med glukose. Løbende måling af blodglukose enten med syre-base-målinger eller med et glucometer anbefales, da der i senforløbet af SE kan opstå hypoglykæmi, som skal adresseres.
Samtidig med monitorering af de vitale systemiske parametre bør der foretages løbende neurologisk vurdering, da langvarigt SE, som sagt resulterer i cerebralt ødem, både cytotoksisk og vasogent (1, 9, 20, 21). En patient i SE, som er i intravenøs antiepileptisk behandling, er vanskelig at monitorere neurologisk, da mange af de normale hjernenervereflekser er dæmpede på grund af behandlingen. Pupilreflekser og pupilstørrelse kan dog altid undersøges. Anisokori, lysstive pupiller med miosis eller mydriasis er et vigtigt varselstegn ved udvikling af hjerneødem. Dette bør adresseres ved at vedligeholde ilt- og blodtilførslen til hjernen på overarbejde, samt ved medicinsk behandling for hjerneødem, herunder IV-behandling med mannitol og glukokortikoid (dexametason anbefales).
Udredning for underliggende årsag
Det er af stor vigtighed, at der i den akutte situation med en SE-patient undersøges for evt. underliggende årsager – ekstrakranielle såvel som intrakranielle, da manglende korrektion af underliggende sygdom er en hyppig årsag til behandlingssvigt (1).
Antileptika |
Rute |
Dosis |
Tmax |
T1/2 |
| Diazepam | IV IN PR | "1 mg/kg hvis ikke allerede i antiepileptisk behandling 2mg/kg hvis i anden antiepileptisk behandling CRI 0,1-0,5 mg/kg/hr" |
"<3 min (IV) 5 min (IN) 15 min (PR)" |
Ca. 3 timer |
| Midazolam | IV IM IN | "0,2-0,4 mg/kg CRI 0,05-0,5 mg/kg/hr " |
<10 min (IV) | 1-1,5 time |
| Phenobarbital | IV (IM) | "Bolus á 5-10mg med 20-30 min. mellemrum Max. 12-24 mg/kg/24h" |
N/A (Fordeling til CNS ses indenfor 20-30 min) | Gennemsnit på 56 timer |
| Levetiracetam |
IV SC |
40-60mg/kg som loading dose. Herefter 20mg/kg q8h | <15 min | 3-4 timer |
Tabel 4. Farmakologisk information om de mest almindeligt anvendte antiepileptika (9, 23, 26-32). TMAX = Tid til højeste serumkoncentration; T½ = Halveringstid; IV = intravenøs; IN = intranasal; PR = per rectum; IM = Intramuskulær; SC = subkutan; CRI = constant rate infusion.
På baggrund af den kliniske og neurologiske undersøgelse samt anamnese og hundens signalement vil man oftest kunne danne sig en primær mistanke. Er patienten allerede kendt med epilepsi, er dette en værdifuld oplysning. Hvis der udover anfald er observeret eller er anamnestiske oplysninger om lateraliserede kliniske symptomer fra forhjernen rettes mistanken primært mod intrakraniel strukturel sygdom som tumor eller andre rumopfyldende processer. Sådanne lateraliserede symptomer kan være cirklen i store cirkler (som hovedregel mod den side i hjernen, hvor en læsion er placeret), lateraliseret proprioceptivt tab (som vil være kontralaterale i forhold til læsionen), unilateral blindhed og/eller adfærdsændringer. Ved inflammatorisk hjernesygdom er de kliniske tegn ofte mere diffuse eller multifokale. Inflammatorisk hjernesygdom kan ikke udelukkes på baggrund af fravær af systemisk inflammation. Der kan på den kliniske undersøgelse og/eller anamnese være begrundet mistanke om ekstrakraniel ætiologi, såsom intoksikation, hepatopati eller anden metabolisk sygdom. Ved mistanke om intrakraniel årsag bør skanning af hjernen og CSF-undersøgelse altid overvejes. Dette afventer dog til den primære anfaldskontrol er på plads.
For ekstrakranielle årsager bør som minimum overvejes muligheden for intoksikation eller metabolisk sygdom, såsom hypoglykæmi, elektrolytforstyrrelser og organsvigt. Til dette formål anbefales parakliniske tests: syre-base-måling (inkl. glukose), hæmatologi, biokemi, elektrolytkoncentrationer, urinanalyse, samt eventuelt drug test. Denne udredning anbefales som et minimum til alle patienter i SE. Hvis patienten allerede er kendt med epilepsi og i antiepileptisk behandling med phenobarbital og/eller kaliumbromid, bør serumkoncentrationen af disse måles for at afgøre, om der kan være tale om for lav serumkoncentration, og i forhold til beregning af den maksimale døgndosis for patienten i den akutte antiepileptiske behandling.
Ved mistanke om indtag af en giftig substans bør induktion af opkast, tildeling af aktivt kul og/eller ventrikelskylning overvejes, men det er ikke altid muligt hos en patient i SE. Er der tale om kutant optagne toksiner (fx insekticider), bør dyret vaskes. Mange stoffer i familiedyrs nærhed kan være krampeudløsende. En liste over disse kan findes i lærebøger eller ved søgning på internettet, men faktum er, at man i klinikken sjældent kender det specifikke stof, der forårsager anfaldene. Der findes tillige en lang række giftige planter, der potentielt kan forårsage anfald (25).
Anfaldskontrol
Efter anlæggelse af intravenøs adgang og den første akutte stabilisering af patienten igangsættes behandling med antiepileptisk medicin. Der er forskellige muligheder for medicinsk anfaldskontrol, og det er vigtigt at behandle denne akutte tilstand med både korttidsvirkende og langtidsvirkende antiepileptika. Det er selvfølgelig også vigtigt i denne sammenhæng, at der er de nødvendige antiepileptika til rådighed i klinikken. Nedenfor gives konkrete anbefalinger vedrørende anfaldskontrol samt farmakologisk information om de mest effektive antiepileptika for SE-behandling (tabel 4).
Benzodiazepin
Benzodiazepiner (BZD), såsom diazepam og midazolam, er potente hurtigtvirkende antiepileptika og derfor førstevalg i den indledende behandling af SE. Deres antiepileptiske virkning foregår gennem binding til og stimulering af den inhibitoriske GABA-receptor, som vist i figur 3. Herved øges influx af chlorid til neuronens cytoplasma, og dette medfører, at cellens evne til at starte et aktionspotentiale nedsættes (hyperpolarisering). Behandlingen er hurtigt indsættende, men imidlertid også korttidsvirkende med en halveringstid for diazepam på ca. 3 timer hos hund. Dette understreger vigtigheden af samtidig behandling med langtidsvirkende barbiturat (se nedenstående). Indenfor 3 minutter efter IV-indgift af diazepam og ca. 8-10 minutter for midazolam ses den højeste serumkoncentration, og fra blodet sker fordelingen af både diazepam og dets potente metabolitter til centralnervesystemet (CNS) hurtigt grundet den høje lipofilicitet (intravenøs diazepam passerer fx blodhjernebarrieren i løbet af 1 minut). Behandling med rektal diazepam er en mulighed, men da der ses dårligere optag og længere tid til terapeutisk serumkoncentration, er IV-behandling at foretrække (28). Hvis IV-adgang ikke er muligt, kan rektal behandling med diazepam gives; alternativt kan intranasal (IN)-behandling med enten diazepam eller midazolam anvendes, hvorved terapeutisk serumkoncentration opnås indenfor 5 minutter (29, 30). IN-behandling kræver dog en speciel forstøver, der kan kobles på en sprøjte.
Dosis af BZD afhænger af, om hunden er i anden behandling; således øges metaboliseringen, og halveringstiden reduceres, hvis patienten er i kronisk behandling med phenobarbital. Som udgangspunkt er dosis for diazepam 0,5-2mg/kg, og 0,2-0,4mg/kg for midazolam, hvor den høje ende af dosis-intervallet kan gives til hunde, som i forvejen er i antiepileptisk behandling med phenobarbital. IV-behandling kan gentages efter 5 min, hvis der ses manglende respons. Ved god effekt af BZD kan constant rate infusion (CRI) overvejes (se tabel 4 og figur 4).
Det er en hyppig årsag til manglende anfaldskontrol, at man fortsætter for længe med benzodiazepiner alene uden at iværksætte samtidig behandling med langtidsvirkende barbiturat (31, 32).
Barbiturater
Ved SE behandling anvendes udover benzodiazepiner også barbituratet phenobarbital (Fenemal/Phenoleptil). Phenobarbital er et meget effektivt antiepileptikum, som i den akutte IV-behandling af SE (modsat BZD) er langsommere om at virke, men til gengæld har en meget længere anfaldsdæmpende effekt. Phenobarbital sætter således ind der, hvor effekten af benzodiazepin svinder, og vedligeholder antikonvulsiv beskyttelse over et længere tidsrum. Derfor er kombinationsbehandlingen med benzodiazepin og phenobarbital så vigtig i forhold til behandlingssucces.
Som BZD binder barbiturater til GABA-receptoren, dog på et andet sted, som illustreret i figur 3. Bindingen til GABA-receptoren medfører forlængelse af åbningstiden for chloridkanalen, influx af chlorid, og forårsager dermed hyperpolarisering af neuronerne. Phenobarbital har desuden en dæmpende effekt på excitatorisk glutamat. Ideelt opstartes behandling med phenobarbital samtidig med BZD for bedst mulig kontrol af SE.
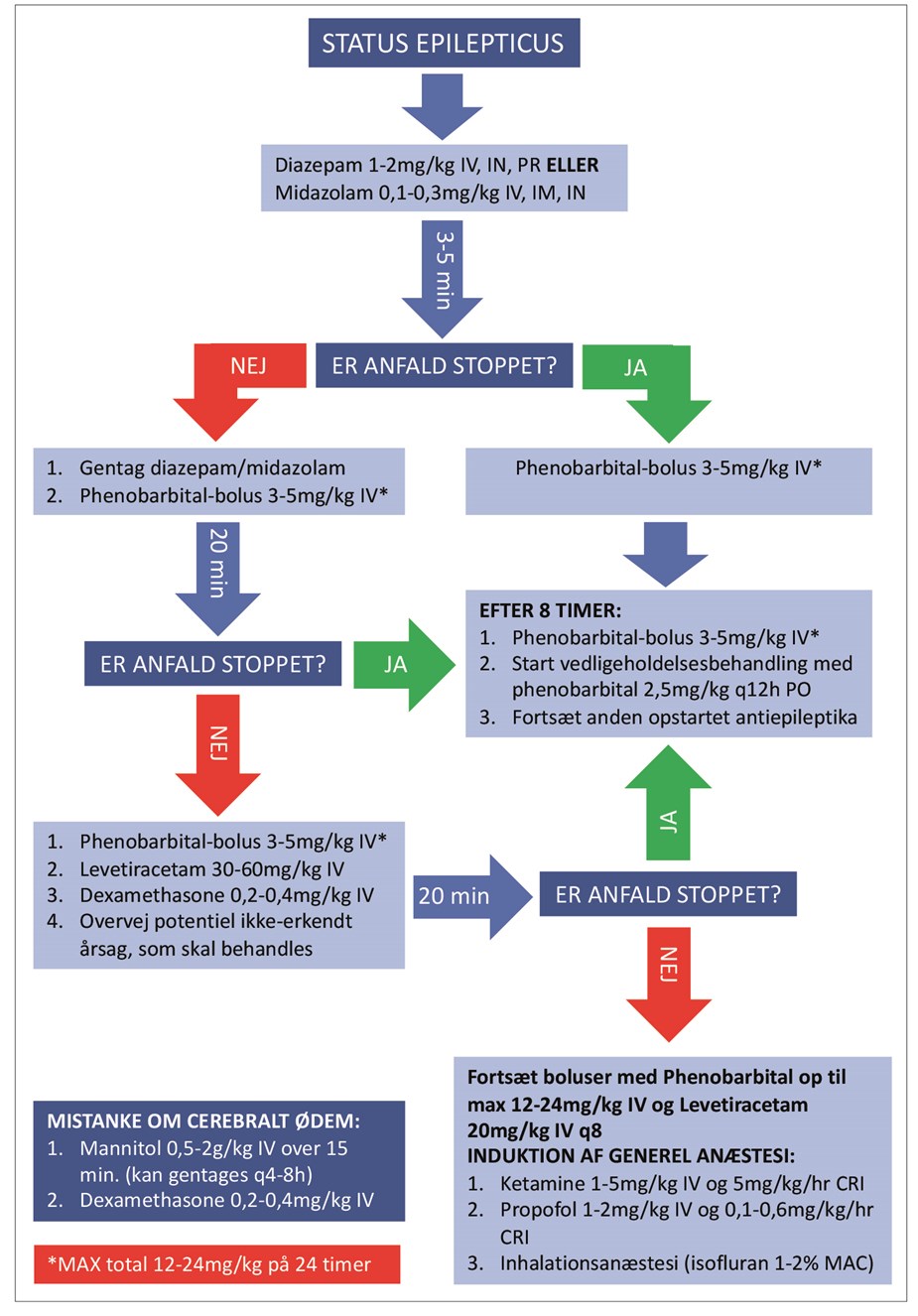
Figur 4. Protokol for generel håndtering af hunde i status epilepticus. Dosis og tidsinterval imellem doseringer bør vurderes fra patient til patient. Protokollen kan ikke erstatte den kliniske vurdering af den enkelte patient. Bemærk desuden, at protokollen udelukkende omhandler anfaldskontrol og ikke systemisk stabilisering, diagnosticering og monitorering af patienten, som er beskrevet i teksten.
Fordelingen til CNS tager længere tid for barbiturater end for BZD grundet lavere lipofilicitet, og således går der 20-30 minutter, før den antiepileptiske effekt af IV-injektionen ses fuldt ud. Til gengæld er halveringstiden markant længere for phenobarbital sammenlignet med diazepam; gennemsnitligt 56 versus 3 timer.
Behandlingen med IV-phenobarbital til SE-patienter bør titreres til effekt. Titreringen kan være en udfordring, da medicinen som sagt fordeles relativt langsomt til CNS og derfor kræver tålmodighed i en situation med markant tidspres. Det anbefales at give behandlingen i mindre boluser over nogle minutter, grundet risikoen for respirationsdepression, især når der behandles med BZD samtidigt (4, 9, 20, 23). Bolusser á 3-5 mg/kg med 20 min. mellemrum anbefales, med et maksimun på 12-24 mg/kg på 24 timer. Se desuden den foreslåede behandlingsprotokol i figur 4.
For SE-patienter, der allerede er i behandling med phenobarbital, findes forskellige metoder til at udregne den optimale dosis phenobarbital. Som tommelfingerregel øges den allerede eksisterende serumkoncentration med 20 mmol/L for hver 3 mg/kg der gives IV (21).
Ved mistanke om hepatisk encefalopati bør benzodiazepiner og barbiturater så vidt muligt undgås. Her kan IV-Levetiracetam anvendes, indtil udredning for underliggende hepatopati er foretaget.
Levetiracetam
Levetiracetam (LEV) har gennem de seneste år været brugt til behandling af SE hos både humane og veterinære patienter (4, 9, 23, 27, 32). Virkningsmekanismen er ikke fuldt klarlagt, men det er sandsynligt, at LEV udøver sin effekt mange steder i hjernen. Det er påvist, at LEV har neuroprotektive egenskaber, og desuden øges effekten af diazepam ved samtidig behandling. Evidens for LEV’s effektivitet i SE-behandling er begrænset, når man kigger på tilgængelige data, men LEV kan potentielt have gavnlig effekt (9, 27, 28). LEV udskilles primært via nyrerne, hvorfor LEV er førstevalg til SE-patienter med hepatopati.
LEV har få bivirkninger og lav toksicitet, og det er derfor et meget sikkert farmaka som add-on- behandling af SE. Halveringstiden hos raske hunde efter IV-LEV er målt til 3-4 timer. I et enkelt studie af Hardy et al., hvor farmakokinetikken af LEV hos 19 hunde er undersøgt, rapporteres en kortere halveringstid hos SE-patienter samt hos patienter i kronisk behandling med phenobarbital (27). Den kliniske betydning af dette er indtil videre ukendt, og der er ikke belæg for ændring af dosis i disse tilfælde.
Behandlingsrefraktær SE
Ved fortsat anfaldsaktivitet og manglende anfaldskontrol (behandlingsrefraktær SE), er det nødvendigt med yderligere tiltag. Propofol kan forsøges som CRI og har sin effekt centralt på både GABA- og NMDA-receptorer. Man bør dog holde sig for øje, at den elektriske anfaldsaktivitet i hjernen kan fortsætte til trods for, at motoraktiviteten stoppes med propofol. Herudover kan der under induktion med propofol og i opvågning til tider ses anfaldslignende symptomer såsom myokloni og opisthotonus. Patienten bør intuberes og overvåges nøje mht. respiratorisk og kardiovaskulær tilstand, da propofol ofte giver apnø, og ventilering er nødvendig (9, 24).
Ketaminbehandling har i flere studier vist at have effekt på SE-tilfælde, som ellers er behandlingsrefraktære 9, 33-35). En bolus på 1-5 mg/kg efterfulgt af 5 mg/kg/hr som CRI anbefales. Ketamin er en NMDA-antagonist og har dermed en teoretisk berettigelse til behandling af refraktære tilfælde, da den excitatoriske NMDA-receptor som tidligere beskrevet opreguleres efter længere tids anfald. I et nyligt retrospektivt studie af Roynard et al. rapporteredes god effekt af IV-ketamin til behandling af behandlingsrefraktær SE blandt 15 hunde (35). Tidligere har man frarådet ketamin til brug hos patienter med forhøjet intrakranielt tryk (9), men en påvirkning af det intrakranielle tryk ved behandling med ketamin har i nyere studier ikke kunnet påvises (35).
Andre mulige medikamenter til behandling af refraktær SE inkluderer dexmedetomidin (36); loading med kaliumbromid enten oralt eller rektalt (23) samt inhalationsanæstesi (8, 23).
Prognose
Prognosen for SE-patienter afhænger af flere faktorer; ætiologi, tidlig indgriben og behandling samt ejers indstilling. En reel mortalitetsrate trods behandling er ikke fastsat, idet en del patienter aflives inden diagnostisk udredning og intensiv behandling, men flere veterinære studier angiver at 25-32 % af hunde med SE eller klyngeanfald bliver aflivet eller dør uagtet og ofte uden kendt diagnose (2, 7). Humant er rapporteret omkring 20 % mortalitetsrate uagtet ætiologi. Der ses dog flere strukturelle årsager til SE humant grundet den højere incidens af disse, herunder stroke (10).
Strukturel epilepsi som årsag til SE har den dårligste prognose (2, 3, 7, 12). Hvor årsagen til anfald er ekstrakraniel er risikoen for at udvikle SE større end for de intrakranielle ætiologiers vedkommende, men prognosen er til gengæld bedre, når den underligende årsag korrigeres (3, 12, 37, 38).
Konklusion
SE er en klinisk kompleks sygdomstilstand, som kræver mange samtidige overvejelser og akut handling på flere fronter. Disse inkluderer opmærksomhed på behandlingsvinduet og udviklingen af SE fra kompensatorisk til dekompensatorisk fase, identifikation af underliggende årsag, behandlingsplan med både korttids- og langtidsvirkende antiepileptika, monitorering af vitale parametre og støttende behandling. Dette foregår simultant, hvilket er en stor udfordring for klinikeren. Derfor er det også en god støtte at følge en standardprotokol som fx den, der er foreslået i nærværende artikel.